

As with everything on our site, we encourage you to check with a doctor that understands nutrition before making this or anything a regular part of your diet.
Author: Michelle Cole (Founder of Chiari Bridges and Worth The Fight)
New Chiari & Comorbid Symptoms Compilation


These are more than just Chiari symptoms, but they are important, because we usually have more than just Chiari. This is a pretty thorough list. It can be a little emotional for some, as it has a lot of symptoms that doctors have dismissed.
- Print out this PDF.
- Go over the list and put a dot next to all of the symptoms that you have EVER experienced
- Go back over the list and put a check mark next to all of the symptoms that you are CURRENTLY experiencing.
- Go back over the list and highlight the ten symptoms that would help you get your life back the most, and start there!
NEW LIST
OLD LIST
Brain Under Pressure – Understanding Intracranial Hypertension [UPDATED]
![Brain Under Pressure – Understanding Intracranial Hypertension [UPDATED]](https://chiaribridges.org/wp-content/uploads/2021/06/IH_AS271511956_resized.jpg)

Intracranial Pressure (ICP) is measured in millimeters of mercury (mmHg). Most scholars agree that on average:
- Intracranial pressures (ICP) between 5-15 mmHg, are considered Normal pressure
- Intracranial pressures (ICP) between 20-30 mmHg, are considered Mild to moderate intracranial hypertension which “requires treatment in most circumstances”,
- Intracranial pressures (ICP) of > 40 mmHg indicates “Severe and possibly life-threatening intracranial hypertension.” [1]
When high intracranial pressure is left untreated, it creates a “pushing effect” towards the only natural escape at the base of the skull (the foramen magnum), and the cerebellar tonsils in the pathway are pushed through the foramen magnum. [2]
Understanding the Monro-Kellie Doctrine (the pressure-volume relationship)
The association between IH/IIH and Chiari Malformation appears to be a malicious intricate pathological circle. The cranium (skull) consists of brain matter, cerebrospinal fluid, and both venous and arterial blood. A hypothesis, referred to as the Monro-Kellie Hypothesis (now better known as the Monro-Kellie Doctrine), states, “The sum of volumes of the brain, CSF, and intracranial blood is constant. An increase in one should cause a decrease in one or both of the remaining two.” Therefore, if an abundance of cerebrospinal fluid (IIH or hydrocephalus), both cranial blood volume and brain matter should be forced to deplete. This depletion is usually directed in the path of least resistance – through the foramen magnum and into the spinal canal. When the brain matter closest to the bottom of the skull (cerebellar tonsils) is pushed through the foramen magnum and into the spinal canal (an Acquired Chiari Malformation), the tonsils act like a cork and blocks the flow of cerebrospinal fluid (regardless of the size of the tonsillar descent), which in turn, continues to raise intracranial pressure.[3]

Venous Hypertension
When an etiological cofactor exists (such as a space-occupying mass), it is considered Secondary Intracranial Hypertension (SIH); when no other cause was identified, it is known as Idiopathic Intracranial Hypertension (IIH) formerly known as Pseudotumor Cerebri. However, recent studies on the connection between Intracranial Hypertension and Venous Hypertension might put an end to the “idiopathic” theory.
Oxygen-rich blood travels from the heart to the rest of the body through the arterial system, then the oxygen-depleted blood returns to the heart through the venous system. We have a host of small veins in our head and they dump into a series of large veins, called sinuses.
Dural Venous Sinus Stenosis occurs when there is a narrowing of one or more of the venous sinuses (most commonly seen in the transverse sinuses or transverse/sigmoid sinus junction, but can also occur anywhere within these sinuses or in the jugular). This narrowing compromises cerebral venous outflow through the jugular vein (stenosis/compression of the jugular vein can also result in elevated intracranial pressure [4]). Transverse Sinus Stenosis (TSS) is most common what was initially assumed to be “Idiopathic Intracranial Hypertension (IIH), formerly known as Pseudotumor Cerebri (PTC). Depending on the study that you are reading, it is proving to be present in 90-100% of IIH patients [5]. While its connection might sound obscure, if you look at it from a Monro-Kellie perspective – the blood going into the head, cannot get out at the same speed (because of the narrowed sinus). When this inflow of blood remains constant and the outflow is hindered, the transverse sinus on that side (we have two transverse sinuses, one on each side) enlarges, forcing the CSF and brain matter to reduce to maintain the volume equilibrium. This reciprocation can happen when any of the sinuses or jugular narrow (stenosis). While scholars continue to debate whether TSS is a cause or consequence of IIH, surgeons continue to decompress us without checking our pressures or decompress (the most invasive treatment) in hopes that it will lower our pressures, and patients are left with untreated high pressure still causing a “pushing down effect” and an enlarged foramen magnum for our brains to be pushed down. [2] The sagging brain once again obstructs the flow of cerebrospinal fluid by plugging the foramen magnum, and that in turn raises the intracranial pressure even more. Or, the untreated high pressure blows through the duraplasty and causes a post-operative leak, known as a pseudomeningocele.
Reducing the Risks of Post-Op IH/IIH Complications by Identifying Potential Problems Early:
Brain MRIs often show indicators of Intracranial Hypertension (IH/IIH), therefore, we recommend that all Chiari patients have full brain MRIs and not just cervical MRIs.
- When the pressure builds inside of the dura mater the pressure pushes the dura and fluid inside of the crevice that holds the pituitary gland (the sella turcica or pituitary fossa). When the amount of fluid is equal to or greater than 50% and the pituitary gland size is 2mm, the condition is known as Empty Sella Syndrome. (Doctors now recognize that < 50% (where the pituitary gland size is 3-7mm) can also cause symptoms and they now refer to that as a partially empty sella.) [8]

- Slit like or flattened lateral ventricles from the increased pressure, however, when the Foramen of Monro (the aqueduct that connects the lateral ventricle to the third ventricle) is stenosed, the fluid will back-up and the lateral ventricle will not appear flattened. [7]
- Enlarged/swollen optical nerves (papilledema). [8]
- Low lying or herniated tonsils (often diagnosed as a Chiari Malformation). [2]
Ideas BEFORE DECOMPRESSION is considered:
If you have symptoms of IH/IIH accompanied by any of the MRI indicators mentioned above, it is both reasonable and prudent to ask your neurosurgeon to investigate further BEFORE DECOMPRESSION.
- See a neuro-ophthalmologist to check for signs of papilledema, including Optical Coherence Tomography and Ultrasonographic B-scanning. [8]
- Magnetic Resonance Venography (MRV, preferably with the ATECO technique) to check for venous stenosis of any of the cranial sinuses and/or jugular vein. Stenosis is not exclusive to the transverse sinus and it can happen in multiple sinuses simultaneously.
- If overweight, consider trying to lose weight. Studies show that a weight loss of 5-10% of one’s overall body weight, when accompanied by a low-salt diet, can offer some to IH/IIH symptoms.[9]
- Consider trying Diamox (Acetazolamide) and/or Topamax (Topiramate) to see if that improves the pressure headaches.
- Request a lumbar puncture (spinal tap) to test your opening pressures. We recommend that it’s guided with fluoroscopy with a small gauge needle (and not the standard 22 gauge) that they allow to drip (as opposed to syringe pull) and ensure that someone is available to perform an epidural blood patch if necessary. Time should be allotted afterward to lay flat for several hours immediately following the procedure and for several days once returning home. The potential for CSF leaks is high for the EDS/Chiari patient. A doctor that marginalizes the risks ahead of time, will generally marginalize your symptoms when you are actively leaking.
- ICP Bolt Monitoring can record the differences experienced in pressure over time, and how different positions affect ICP.
Note: When the intracranial pressure gets high enough, it can cause a cranial leak. This is especially true for the Ehlers-Danlos patient where the dura mater is thin and fragile. When a cranial leak decreases the intracranial pressure, the papilledema, empty sella, stenosis, and high-pressure headaches can sometimes start to revert to normal or near-normal, and the leak will affect any attempts to check intracranial pressure (reducing the pressure from what it was before the leak occurred), however, the tonsillar herniation will usually remain if the pressure gets too low. [10]
TREATMENT OPTIONS:
- If you have IH/IIH, ask your doctor to help you reduce the ICP before decompression! This can be done medicinally using Topamax or Diamox. If that doesn’t work, a shunt or stent should be considered.
- A Ventriculoperitoneal Shunt(VP Shunt) or Ventriculoatrial Shunt (VA Shunt) can be surgically placed to drain cerebrospinal fluid straight from the ventricle. Shunts are known for failing and often need a multitude of revisions, but even with all the revisions, it can be less invasive than a decompression. Shunts under the foramen magnum should never be used as a means of controlling ICP.
- An MRV is recommended for all IH/IIH patients. If it indicates venous stenosis, stenting should be considered, as leaving the sinus/jugular stenosed can post other health risks, and stenting can be successful for many with fewer complications that often require revisions.
For the IH/IIH patient, herniated tonsils should be assumed an Acquired Chiari Malformation (even if a small posterior fossa is evident), and by correcting the high pressure before decompression, the decompression will be less likely to fail.
Helpful Tips:
If you have IH/IIH, it is best to avoid caffeine, avoid progestin based birth control, and all EDS patients should try to avoid the use of fluoroquinolones such as ciprofloxacin (Cipro), levofloxacin (Levaquin/Quixin), gatifloxacin (Tequin), moxifloxacin (Avelox), ofloxacin (Ocuflox/Floxin/Floxacin), norfloxacin (Noroxin), due to the increased risk of aneurysm.
References:
1 Rangel-Castillo, Leonardo, et al. “Management of Intracranial Hypertension.” Rangel-Castilla, Leonardo et al. “Management of intracranial hypertension.” Neurologic clinics vol. 26,2 (2008): 521-41, x. doi:10.1016/j.ncl. Feb. 2008, <https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2452989/>.
2 Aiken, A.H., et al. “Incidence of Cerebellar Tonsillar Ectopia in Idiopathic Intracranial Hypertension: A Mimic of the Chiari I Malformation.” American Journal of Neuroradiology; Nov. 2012, <http://www.ajnr.org/content/33/10/1901>.
3 Mokri, B. “The Monro-Kellie Hypothesis: Applications in CSF Volume Depletion.” Neurology., U.S. National Library of Medicine, 26 June 2001, <https://www.ncbi.nlm.nih.gov/pubmed/11425944>.
4 Zhou, D., et al. “Intracranial hypertension induced by internal jugular vein stenosis can be resolved by stenting.” European Journal of Neurology, November 2017 <https://onlinelibrary.wiley.com/doi/abs/10.1111/ene.13512>.
5 Henderson, Fraser C., et al. “Neurological and Spinal Manifestations of the Ehlers–Danlos Syndromes.” American Journal of Medical Genetics Part C: Seminars in Medical Genetics, 21 Feb. 2017, <www.onlinelibrary.wiley.com/doi/10.1002/ajmg.c.31549/full>.
6 Pietrangelo, Ann. “Empty Sella Syndrome.” Healthline, Oct. 2017, <https://www.healthline.com/health/empty-sella-syndrome>.
7 Hingwala, Divyata R., et al. “Imaging signs in idiopathic intracranial hypertension: Are these signs seen in secondary intracranial hypertension too?.” Annals of Indian Academy of Neurology vol. 16,2: 229-33. doi:10.4103/0972-2327.112476, June 2013, <https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3724081/>.
8 Mollan, Susan P., et al. “A practical approach to, diagnosis, assessment and management of idiopathic intracranial hypertension.” Practical neurology vol. 14,6: 380-90. doi:10.1136/practneurol-2014-000821. May 2014, <https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4251443/>.
9 Thurtell, Matthew J., and Michael Wall. “Idiopathic Intracranial Hypertension (Pseudotumor Cerebri): Recognition, Treatment, and Ongoing Management.” Current Treatment Options in Neurology, U.S. National Library of Medicine, Feb. 2013, <https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3554852/>.
10 Pérez, Mario A., et al. “Primary spontaneous cerebrospinal fluid leaks and idiopathic intracranial hypertension.” Journal of neuro-ophthalmology : the official journal of the North American Neuro-Ophthalmology Society vol. 33,4: 330-7. doi:10.1097/WNO.0b013e318299c292, Dec. 2014, <https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4040082/>.
The Important Questions to Ask Your Neurosurgeon [Revised]
Most Chiarians go to see a surgeon with an expectation of them being knowledgeable in their field. However, while they might be a neurosurgeon, their knowledge of Chiari and its comorbid/pathological conditions might not rank high in their practice. Make the most of your initial appointment by interviewing them and what they really know about Chiari Malformations. Be cautious of inflated success rates. Chiari decompression in general offers a just over a 50% success rate (which means it has a nearly 50% failure rate). Surgeons that claim a 100% (or near 100% success rate) are usually not basing their success on how their patients feel afterward, it is based on if they were successful with the aspects of the surgery:
Removal of the occipital bone ✓
Opening the dura and adding the patch/graft ✓
Laminectomy ✓
Cauterization/resection of cerebellar tonsils ✓
WE DESERVE BETTER THAN THAT!
HERE IS A LIST OF CHIARI QUESTIONS WE RECOMMEND ASKING AT YOUR FIRST NEUROSURGERY APPOINTMENT:
General Questions:
- How do you define a Chiari Malformation?
- What do you believe causes a Chiari malformation?
- Are all Chiari malformations from a small posterior fossa?
- Do I have a small posterior fossa? If yes, how big is it? If size is unknown, was my posterior fossa measured? If not, why not? How did you come to the conclusion that I have a small posterior fossa?
- How common do you believe Acquired Chiari malformations to be?
- Do you always recommend decompression surgery for all of your patients with herniated cerebellar tonsils? Why/why not?
- In an average month, how many Chiari decompressions do you perform? How many tethered cord releases? How many craniocervical fusions? What percentage of your practice is spent treating patients with these connective tissue related conditions?
- Looking at my brain scan, is any part of my “brainstem” herniated (below the posterior fossa)? If so, does that make me a Chiari 1.5?
Intracranial Hypotension (low pressure) Questions:
*Article to help you understand CSF Leaks & Intracranial Hypotension prior to your appointment.
If you have SYMPTOMS OF LOW INTRACRANIAL PRESSURE and/or suspect a cerebrospinal fluid leak, we recommend asking the following questions:
- S.E.E.P.S.
- Looking at my brain scan, do you see any Subdural fluid collections?
- Looking at my brain scan, do you see an Enhancement of pachymeninges?
- Looking at my brain scan, do you see an Engorgement of my venous structures? Should we do an MRV to make sure?
- Looking at my brain scan, does my Pituitary appear to be enlarged?
- Looking at my brain scan, does my brain appear to be Sagging?
- Looking at my corpus callosum:
- Does there appear to be a depression?
- Is there an inferior pointing of the splenium?
If he/she answers affirmatively to any of the above S.E.E.P.S. questions, ask:
- What should be done to find/repair a potential leak?
- Are you aware that it is common for CSF Leaks to not show up on MRI?
- Are you willing to do a CT Myelogram and/or a digital subtraction myelogram, if I develop symptoms of a leak and none can be found on MRI?
- Are you aware that it can often take multiple epidural blood patches to try and seal a leak, and sometimes when a blood patch fails to work, a surgical dural repair might be necessary?
Intracranial Hypertension (high pressure) Questions:
*Article to help you understand Intracranial Hypertension prior to your appointment.
If you have SYMPTOMS OF HIGH INTRACRANIAL PRESSURE, we recommend asking the following questions:
- Looking at my brain scan, do I have cerebrospinal fluid in my sella turcica (Empty Sella Syndrome)?
- Looking at my brain scan, do you see any evidence of my optic nerves are swollen (papilledema)?
- If so, should I be referred to a neuro-ophthalmologist?
- Looking at my brain scan, do my lateral ventricles appear small or flattened?
- If so, do I need to have my pressures checked?
- If yes, are you aware of the risks of developing a CSF Leak from a lumbar puncture?
- What are the symptoms of a CSF Leak, should one develop?
- What is your plan of action if I should develop these leak symptoms?
- Are you aware that it is common for CSF Leaks to not show up on MRI?
- Are you willing to do a CT Myelogram if I develop symptoms of a leak, and none can be found on MRI?
- Should a leak be found, are you aware that it can often take multiple epidural blood patches to try and seal a leak?
- If so, do I need to have my pressures checked?
Tethered Cord Questions:
*Article to help you understand Tethered Cord: Sorry, Coming Soon.
If you have SYMPTOMS OF TETHERED CORD, we recommend asking the following questions:
- Looking at my brain/cervical scan, does my brainstem appear to be elongated?
- Looking at my cervical scan, does my spinal cord appear to be stretched?
- Looking at my lumbar scan, does my conus reach my mid/low L2?
- Looking at my thoracic and lumbar scan, does my spinal cord appear to be pulling to the back, or one particular side?
- If so, should we do a prone MRI to see if it has actually adhered to that side?
- Looking at my lumbar scan, do I appear to have fatty tissue inside the epidermis?
- If the answer to any of these questions is affirmative, do you suspect that I have a tethered spinal cord?
- If so, should we plan for a Tethered Cord Release before or soon after decompression surgery, so the likelihood of a failed decompression is reduced?
- If I have urological issues, can I get a referral for urodynamic testing to rule out any other potential causes of my urological issues?
Craniocervical Instability (CCI) & Atlantoaxial Instability (AAI):
*Article to help you understand CCI & AAI prior to your appointment.
If you have SYMPTOMS OF CRANIOCERVICAL INSTABILITY or SYMPTOMS OF ATLANTOAXIAL INSTABILITY, we recommend asking the following questions:
- Looking at my brain/cervical scans, what are the measurements of my clivoaxial angle, Grabb-Oakes, and Harris Measurements?
- Do these measurements meet the diagnostic criteria for Craniocervical Instability?
- Looking at my flexion and extension imaging, how many millimeters of translation are there between flexion and extension?
- Does Chamberlain’s Line cross my odontoid? If so, does it cross at a level that would indicate Basilar Invagination?
- Looking at my rotational imaging, what is the percentage of uncovering of the right and left articular facets on rotation?
- Do the percentages from my rotational imaging meet the diagnosis criteria for Atlantoaxial Instability?
IF A DIAGNOSIS CRITERIA IS MET IN ANY OF THE ABOVE, WE STRONGLY RECOMMEND THAT YOU WAIT ON DECOMPRESSION AND PURSUE THE TREATMENT OF SAID CONDITION(S) AND THAT OF EHLERS-DANLOS SYNDROME, AS EACH OF THESE CONDITIONS CAN BE PATHOLOGICAL TO AN ACQUIRED CHIARI AND EACH IS A STRONG INDICATOR THAT A CONNECTIVE TISSUE PROBLEM EXISTS.
*The questions in this article will periodically change as we are able to expand our recommended questions.
*Original version released September 2018, revised 2023.
Struggling Through the Holidays

‘Twas the night before Christmas and despite the sleeping spouse, there was still one stirring in the Chiarian’s house. The stockings were hung by the chimney with care as she hoped despite the pain, she’d be able to be there. The family was nestled all snug in their beds, while fear of disappointment danced through her head.
While everyone’s talking about holiday cheer and how there’s laughter in the air, for the chronic pain patient it’s not that easy to get into the holiday spirit. We remember the happier holidays of the past and all that people want to see in us, but there are so many thoughts acting as obstacles in our path.
Will I have the spoons (energy) that I need to make it through the day?
We speak of energy in terms of spoons (The Spoon Theory, by Christine Miserandino). We know what it was like to have normal levels of energy to accomplish tasks and how much more energy every task requires now that our bodies went crazy. You don’t appreciate the energy it takes to get ready for something until you need a nap after every shower you take.
What will I do if I experience a pain flare and how will everyone else respond to me if I do?
Almost worse than the pain itself is living in fear of the pain, especially when we know how it seems to ruin everything for everyone, not just us. For the patient and their family, they know far too well how pain can ruin even the most important of occasions. And for the patient, we know the look on the faces of those we love when we have to cancel or depart early. It’s one thing to see those faces a time or two in a lifetime, but it’s a lot harder when it happens time and time again, and there’s nothing you can do about it.
Will I be able to engage?
People rarely realize how much time we really spend alone (or at least alone in our thoughts). We think about so many things. Should I tell them about what I’m facing? Should I answer how I’m really feeling, or just say, “I’m fine”? Am I talking too much about my conditions? Is it just me and my brain, or is it them? Are my feelings about this even rational? Am I losing my mind? Most of these thoughts are actually healthy thoughts, but when we second guess engaging with the world and live in constant fear of offending, it becomes detrimental to the way we see our value on this earth.
How many days of pain will I experience after the holiday is over?
We’ve learned from those times that we’ve tried to “push through the pain,” that this will be a factor nearly 100% of the time. While the healthier us could push through the pain, that often backfires when it comes to chronic pain. After a few hours of festivities (no matter how light the festivities seem to be), our nervous systems usually respond to the stress with inflammation and pain (which can last several days or even weeks).
Will I live up to what’s expected of me or am I going to let down everyone I love, yet again?
Even when nobody around us expects much from us, there’s always a part of us that still longs to be like our former selves – to have the strength and energy that we once had. The truth is, despite everything we’ve been through, we want to be more for those that we love. Our lives were forever altered and reconciling that with a lifetime of dreams isn’t easy. We’re not feeling sorry for ourselves, we’re mourning and trying to adapt to the reality of all that we face. It hasn’t been easy on our families either, they’re in mourning too. Chiari/comorbids have stolen hopes and dreams from all of us, but we don’t have to let it dominate us. We can figure it all out together and be a stronger family for it!
September Is Chiari Awareness Month

It’s hard having a chronic illness that isn’t all that understood.
As patients, we have to fight on absolutely every level!
Before diagnoses, we fight for someone to hear us when:
- We explain to them that our neck is to weak to hold up our head.
- We’re trying to hold our heads up with our hands when laying back isn’t an option.
- Our necks start spasming to the point that we feel like we’ve been internally decapitated.
- We have to ride in the front seat to try and minimize the car sickness.
- We suddenly can’t balance to walk.
- Our eyes start twitching beyond what could ever be considered normal.
- We aren’t able to do what we could just a short time ago, or even a few hours ago.
- That we want to scream and cry because of the pain, but we know it will only make it worse.
- We go to say something and can’t find the right word because it just isn’t in our memory bank at that moment.
- We spontaneously can’t read because we have double vision, blurred vision, or our eyes wont stop jerking around to focus, yet an hour later we’re fine.
- We explain that doctors not knowing what’s wrong doesn’t mean that nothing is wrong (even when they say nothing is wrong).
Around diagnoses, we fight to:
- Process the magnitude of what we’re facing.
- Learn all we can so we’re prepared for the important decisions before us.
- Find the right doctors who are knowledgeable and trained in our condition(s).
- Fight to be stoic when we know that it’s not just our bodies enduring all of this – something is breaking in our souls and we’re fighting to not let it change us for the worse.
When our doctors continue to dismiss our symptoms, we need our friends and families to understand:
- That we’re still the same wife/husband, mother/father, sister/brother, aunt/uncle, and/or friend that you’ve known and loved for all these years, and we need you now more than ever!
- That decompression is not a cure! In fact it typically fails to relieve symptoms over the long term nearly 50% of the time when pathological conditions aren’t treated beforehand.
Testimonials

You must be logged in to submit this form.
WTF! Online Support Group Meetings

Ever feel alone in all you’re facing? It’s one thing to read what people are going through, but to see them as they share how they’re going through the same struggles that you are can be game-changing!
Starting in December, the tribe will be hosting online support meetings on the second and fourth Wednesday of each month to share and learn about all we’re dealing with.
A valuable resource for patients and their families, looking for support and a sense of direction!
The Prayer of An EDS/Chiari/Comorbid Patient


As we lift up a warrior fighting EDS, Chiari, and/or Comorbids,
We are believing You for:
Knowledgeable doctors/surgeons
With hearts for the patients that are trusting them
Ears to hear them
And a willingness to unlearn and relearn
CSF leaks to seal
Cranial masses to disappear
Narrowed venous structures to widen
And cranial pressures restored to normal
Sticky filums to release
Stretched spinal cords to retract
Conus Medullaris’ to rise
And elongated medullas restored without consequence
Collagen restored without mutation
Intravertebral discs moving back into their rightful place
Laxity issues resolved
Straightened odontoids and clivus bones
And craniums to rise
Spines to straighten
Cerebellar tonsils to rise
CSF flow restored
And syringes (syrinxes) dissipated
Muscles reconditioned
Paralysis reversed
Vision completely restore
Ringing in the ears to cease
Habitual good night’s rests
No insomnia, painsomnia, chronic fatigue, or narcolepsy
Breathing issues corrected
Restless legs calmed
Nerves decompressed
Even vagus restored
Motility perfected
Inflammation gone
And pain a thing of the past
Depression replaced
Hearts seasoned with grace
Families restored
Where no one’s needs are ignored
Thank You, Father,
That despite all we’ve endured,
Your grace continues to be sufficient.
That none of this has taken You by surprise;
You knew all we’d face, yet You still chose each of us and call us Yours.
Our hope is in You and You alone,
The Author and Perfector of our faith,
We stand on Your promise that You still have a plan for each of us!
We might not know what it is or understand how we’ll get there,
but You are a good Father, and we trust You completely.
In Jesus’ Name, Amen!
(Note: This prayer was written to become a collaborative prayer, that we can add to as needed.)
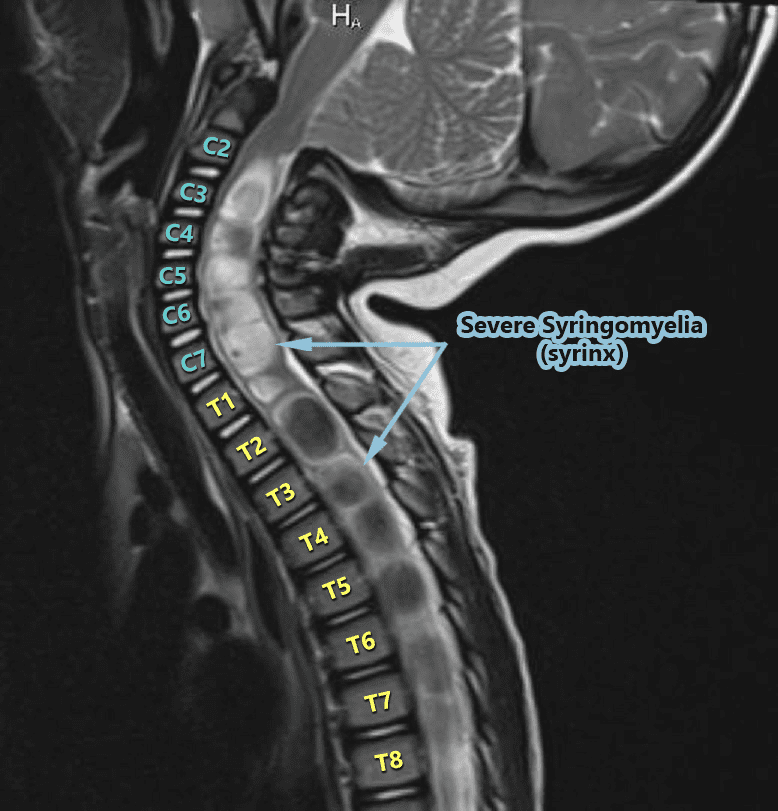
From The Inside Out – Understanding Syringomyelia & Syringobulbia

WHEN A DOCTOR TALKS ABOUT THE “MOST SERIOUS” COMPLICATIONS SURROUNDING CHIARI MALFORMATION, THEY USUALLY SPEAK OF PARALYSIS OR DEATH. WHILE BOTH OF THESE ARE FAR LESS COMMON THAN THE ARRAY OF OTHER SYMPTOMS AND COMPLICATIONS, THEY BOTH CAN INVOLVE THAT OF A SYRINX.
The word syrinx (seer-inks), plural syringes (seer-en-geez), means cavity or cyst. Syringomyelia (seer-ingo-my-el-lee-uh) is when the cyst forms in the spinal cord (myelo usually refers to the spinal cord), and when the cyst is in (or ascends up into) the bulbar region of the brainstem (the medulla oblongata) it is called Syringobulbia (seer-ingo-bulb-e-uh). While these cysts are technically the same cerebrospinal fluid-filled cysts, because they are damaging a different part of the body (each with a completely different function), the location of the syrinx has everything to do with the symptoms that it can cause.
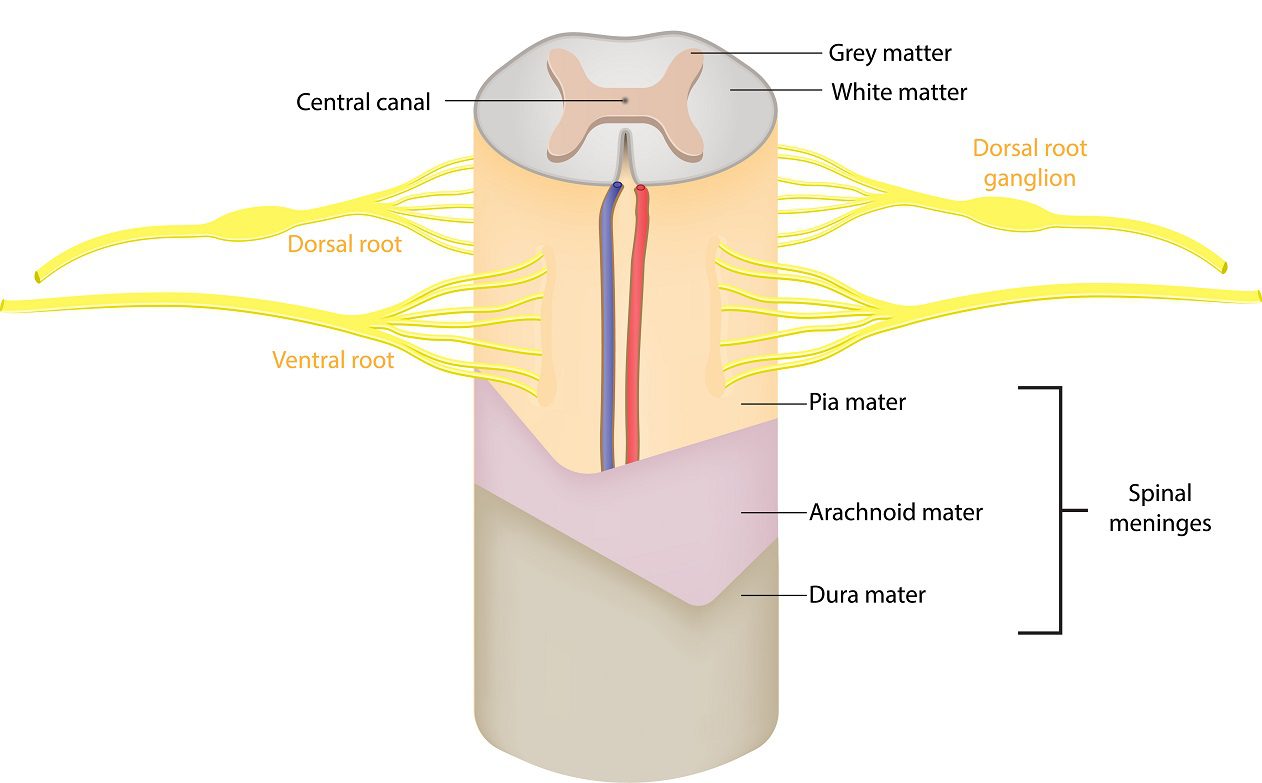 Hydromyelia:
Hydromyelia:
At the center of the spinal cord, there is a hole known as the central canal which runs from the fourth ventricle down the length of the cord. Its role is to carry a microscopic amount of cerebrospinal fluid down the center of the cord from the fourth ventricle. The small amount of fluid in the central canal is usually considered normal. When the central canal has too much cerebrospinal fluid coming in from the fourth ventricle it widens the canal and that abnormal widening that follows the central canal is known as hydromyelia (hydro-my-el-lee-uh) or syringohydromyelia (seer-ingo-hydro-my-el-lee-uh).
The Central Nervous System (CNS) is an amazingly complex network. The brain, brainstem, and spinal cord each play a role in communicating to coordinate everything that happens in our bodies (even when no cognitive thought is required). The messages to and from the brain are sent through the brainstem, down through the spinal cord, and out to our nerves. When that signal is blocked, it can cause interruption from that point down. When it’s thin, it interrupts the signal in the middle (gray matter), as it widens it interrupts the signal more and more, so it’s harder to get signal through the cord at the widest point of the syrinx.
 The spinal cord runs through a canal known as the spinal canal. The spinal canal holds cerebrospinal fluid that among other things, nourishes the spinal cord and helps protect it from injury. A syrinx is generally known to be caused by a blockage of cerebrospinal fluid, which is why it is most commonly seen amongst those with Chiari Malformation. When the cerebellar tonsils descend into the spinal canal, like a cork the tonsils block the flow of cerebrospinal fluid. Even when the tonsils fail to descend, they can rest on top of the foramen magnum and still block the flow of cerebrospinal fluid (see Chiari Zero below). There are other conditions that can cause similar blockages as well, so it is not exclusive to Chiari malformations. Anything that causes stenosis (narrowing) of the spinal canal (the cord: canal ratio) can also block cerebrospinal fluid and lead to the formation of a syrinx – conditions such as bulging/herniated discs (from degenerative causes or trauma), spinal cysts/tumors, edema/inflammation of the spinal cord or surrounding membranes (from trauma or conditions like meningitis/arachnoiditis), and/or irregular curvatures of the spine (scoliosis).
The spinal cord runs through a canal known as the spinal canal. The spinal canal holds cerebrospinal fluid that among other things, nourishes the spinal cord and helps protect it from injury. A syrinx is generally known to be caused by a blockage of cerebrospinal fluid, which is why it is most commonly seen amongst those with Chiari Malformation. When the cerebellar tonsils descend into the spinal canal, like a cork the tonsils block the flow of cerebrospinal fluid. Even when the tonsils fail to descend, they can rest on top of the foramen magnum and still block the flow of cerebrospinal fluid (see Chiari Zero below). There are other conditions that can cause similar blockages as well, so it is not exclusive to Chiari malformations. Anything that causes stenosis (narrowing) of the spinal canal (the cord: canal ratio) can also block cerebrospinal fluid and lead to the formation of a syrinx – conditions such as bulging/herniated discs (from degenerative causes or trauma), spinal cysts/tumors, edema/inflammation of the spinal cord or surrounding membranes (from trauma or conditions like meningitis/arachnoiditis), and/or irregular curvatures of the spine (scoliosis).
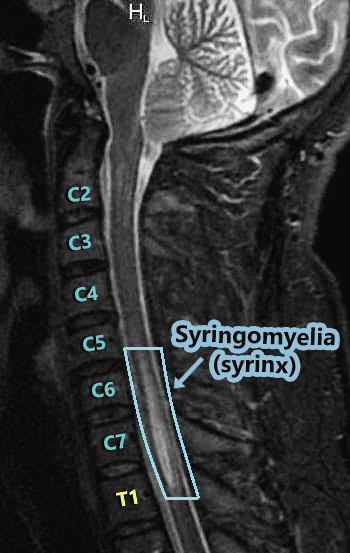 Syringomyelia:
Syringomyelia:
Syringomyelia does not usually run the entire length of the spinal cord like the central canal (but it can). Instead, it often seems to spontaneously appear. As the CSF increases in a syrinx, it can either lengthen or widen. The length doesn’t matter much (radiologists note the location because they report what they see and try to be precise). But a neurosurgeon that knows about syringes, knows that what really matters is the diameter of a syrinx (which is where the risk of paralysis can come into play). Symptoms tend to vary based on where the syrinx is located in the spinal cord. The highest point of the syrinx (where the initial disruption starts) and the widest point of the syrinx (location of the greatest disruption) should always be considered because there are different nerves branching from the spinal cord at different intervals. A syrinx interrupts the communication from the top of the syrinx down, so a syrinx that is higher in the spinal cord can have an impact on a larger range of the body. Someone with a syrinx in the lumbar region of the spinal cord will usually have communication problems from the waist down, but someone with a cervical syrinx is likely to have problems from the neck down. A syrinx in the upper thoracic spinal cord (or above) will often include the arms since the peripheral nerves that lead to the arms branch off around the T1 vertebra. The wider the syrinx, the greater the interruption of communication. Therefore, a syrinx should always be gauged by its diameter and not its length. Symptoms generally include (from the syrinx down): muscle weakness, pain, and spasms in legs; pain, tingling, burning of arms; muscle wasting (atrophy); loss of reflexes; loss of pain senses, loss of temperature sensation, numbness, pain, and stiffness in back/shoulders/upper chest (cape-like area); stiffness of muscles; muscle contractions (fasciculations); bowel & bladder dysfunction; scoliosis; paralysis (rare).
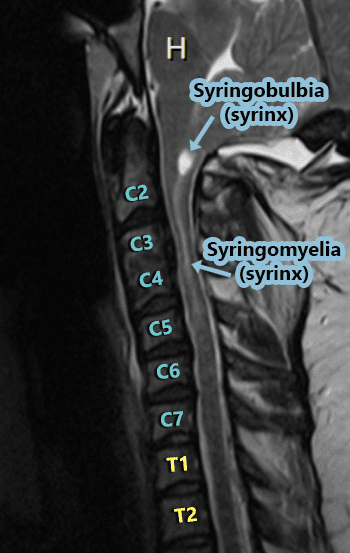 Syringobulbia:
Syringobulbia:
Syringobulbia exists when a syrinx forms in the medulla oblongata (the bulbar region of the brainstem) OR when a syrinx in the cervical cord extends upward into the medulla. A syrinx in the medulla can cause medullary issues or in extreme cases, death (since the medulla is responsible for things that happen autonomically (automatically) for survival – such as breathing, heart rate, swallowing, gag reflex, etc.). Dysautonomia is an umbrella term used to describe any dysfunction of the Autonomic Nervous System (ANS), both Sympathetic and Parasympathetic divisions, often accompany any damage to the medulla. When talking about Dysautonomia, many tend to think of Postural Orthostatic Tachycardia Syndrome (POTS), but POTS is but one symptom of Dysautonomia in a long list. The damage from Syringobulbia is not generally isolated to the medulla, but to the cervicomedullary junction (where the cervical spine meets the medulla), it can also affect the cranial nerves causing symptoms such as facial sensory loss (unilateral or bilateral); extraocular muscle palsy; nystagmus; palatal palsy; atrophy of the tongue; dysphonia (vocal cord paralysis); slurred speech; indistinct speech; drooling; tongue fibrillation; oropharyngeal dysphagia; impaired gag reflex; hearing loss; tinnitus (ringing in the ears); alveolar hypoventilation; Sleep-Disordered Breathing (SDB); Central and obstructive sleep apnea; Anhidrosis (inability to sweat normally); Inability to burp (Retrograde Cricopharyngeal Dysfunction – RCP-D).
Common Treatment Options:
 Monitoring The Syrinx:
Monitoring The Syrinx:
Both Syringomyelia and Syringobulbia tend to be progressive, but in some cases, patients report having no symptoms and imaging proves it to be relatively stable in size. In this case, monitoring is generally recommended. A neurologist or neurosurgeon should carefully monitor these patients to track changes in the diameter of the syrinx (which should include regular imaging) and/or any evolution in related symptoms.
Surgical Treatment Of The Underlying Cause:
For symptomatic patients, or when the syrinx is progressing in diameter, or when the syrinx is so wide in diameter that it is stretching the diameter of the spinal cord from the inside out, treatment is essential. This generally involves treating the cause of the blockage of cerebrospinal fluid. When syrinx exists in a symptomatic patient with Chiari Malformation, a posterior fossa decompression surgery is usually recommended, with the desired result being to re-establish the flow of the cerebrospinal fluid to the spinal canal (so it no longer reroutes to the spinal cord and/or low-lying medulla).
Surgically Draining The Syrinx:
A surgical shunt is commonly used to surgically treat a syrinx when: the underlying cause is unknown or when treating the underlying cause has proven ineffective at reducing the size of the syrinx in a patient that is symptomatic.