
The Michelle Cole Story – A Chiari Warrior’s Journey [UPDATED]
![The Michelle Cole Story – A Chiari Warrior’s Journey [UPDATED]](https://chiaribridges.org/wp-content/uploads/2018/12/DSC07853t.jpg)
As I sit down to update my journey, I am crushed that we’re still figuring things out (and nothing really was as I was initially told it would be), yet at the same time, I’m so thankful that we’re continuing to figure things out. Nobody should have to fight a fight like this (every symptom, every diagnosis), but all of this just increases my resolve to change it before anyone else in my family (or yours) is having to fight it! What we fail to change in our generation, our children and their children will face in theirs!
Looking back, I have always had symptoms of Ehlers-Danlos Syndrome (EDS). As a child, I was in the school nurse’s office for stomach problems at least once a week. I was “double-jointed” and my friends always asked me to do maneuvers that I thought everyone should really be able to do if they tried. I was athletic early on, a tom-boy. I particularly loved playing softball (or baseball with tennis balls was the absolute best), but my ankles rolled when I started to run. Despite the fact that I was the only player that twisted their ankle multiple times in every game, I didn’t think there was really anything abnormal about me. Later, as an adult, I had repeated miscarriages and complications in all of my pregnancies, but the doctors conveniently came up with different explanations for each “rare occurrence.” It couldn’t possibly be all those rare mishaps, but EDS explained it all.
My mother passed away from a brain aneurysm the day after my 18th birthday. She was just 37 years old when she died. As a child she had a lazy eye and scoliosis of the spine, so an eye patch and back brace were a normal part of her childhood attire. She suffered from migraines throughout her adulthood, but nothing was more tale-telling than reading her journal after she passed, with multiple entries about repeated headaches and neck pain. Decades after her death, my maternal grandmother (my mother’s mother) developed multiple brain aneurysms over the course of a decade. Each time one appeared, she had it filled with titanium coils. I always admired her fight for life.

The first headaches that I remember started immediately after giving birth to my first son in 1992. It was a cesarean section at an Army hospital in Fort Ord, California. Instead of an epidural, they gave me three spinal injections to numb me from the chest down. At my postpartum check-up, I complained of daily headaches when upright. My primary care doctor ordered a CT scan, but because it was just a few years after my mom had died they looked only for brain aneurysms and found none. I was still having those orthostatic headaches six months later.

The Accident that Shook Everything
In 2000, I was a Bible College student and stay-at-home mom of three happy and active children (ages 8, 5, and 2). One September night, I was in a car accident that changed all of our lives. My neck was never the same again. My initial symptoms were head/neck pain, but all radiology reports indicated that everything was “unremarkable.” I tried everything they offered to me: rest, acupuncture, acupressure, steroid injections, osteopathic and chiropractic care, nerve stimulation units, physical therapy, pain meds, etc. Nothing worked long-term. Then in 2005, my neurological problems started intensifying. I began having bouts of partial paralysis in my legs and hands. I would just wake up one morning and out of the blue, I would have no fine motor skills. I would wake up feeling as though I had no thigh muscles to support me when I walked or tried to step up a step, and I had difficulty coordinating my footsteps. My primary care doctor at the time did blood tests and concluded that my “potassium level was on the low side of normal, so it must have been from potassium shock,” and he thought that no other tests were warranted. I started having vertigo whenever I was at any elevated height, even just a step or two up, like my brain couldn’t figure out how to balance with visual changes in height (I’d take a step up or down like the step was much higher or lower than it actually was). I also started having noticeable memory issues and intermittent trouble processing information. They tested to see if I was having small seizures in my sleep. When that was ruled out, they referred me to the memory clinic for further cognitive testing. They had no cognitive baseline to compare my results to, but said that I “tested higher than 89% of the population, so I should be happy,” and that I should just try reducing stress in case it was stress-related. They didn’t understand that it didn’t matter to me “how I compared to others.” I was only 34 years old and something was very wrong with me; I wanted answers that had nothing to do with the general population. In 2006, my eyes started twitching all day, every day, until the muscles just wore out and I could no longer hold them open completely. Oddly, one of my college professors inquired about my eyes and recommended that I have it investigated because it “could be neurological in origin.” When I did talk to my doctor about it, he saw the recommendations of the Memory Clinic and attributed it to stress as well, without any testing.

My Chiari Diagnosis
Finally, in 2010, ten years after the car accident, another MRI was done at my insistence to check for aneurysms once again (because I still was having excruciating head/neck pain and trouble holding my head up). I received an email from my primary care doctor that they found a cause of all of my symptoms. It was a condition called Chiari Malformation and the neurosurgery department would be contacting me to make an appointment. The neurosurgeon (who became my neurosurgeon) checked through my MRIs and said that the Chiari Malformation was evident on my first MRI after the accident ten years earlier. I was told that it was congenital and that it is commonly believed to be a result of prenatal drug use or lack of proper prenatal care (which was devastating to hear, but not all that unlikely as I was born in 1971. It also ended up being very wrong “textbook information” that they tell us all). Desperate for a measure of relief, I underwent a full decompression surgery a few weeks later. Missing the fact that part of my brain was in my spinal canal was 100% the hospital’s fault, but in hindsight, I really wish that I had done more research before surgery. I had comorbid conditions (many of which my doctors hadn’t even heard of, didn’t fully understand, and more importantly, they didn’t know the connection between these comorbids and my herniated tonsils). Initially, I felt quite a bit better. The release of pressure in my head helped my headaches. It was short lived though. Those undiagnosed comorbids caused my decompression to ultimately fail, although it all unfolded over several years.
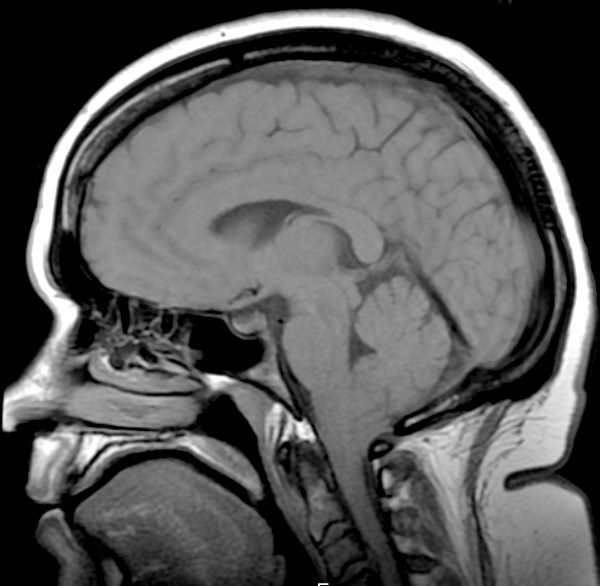
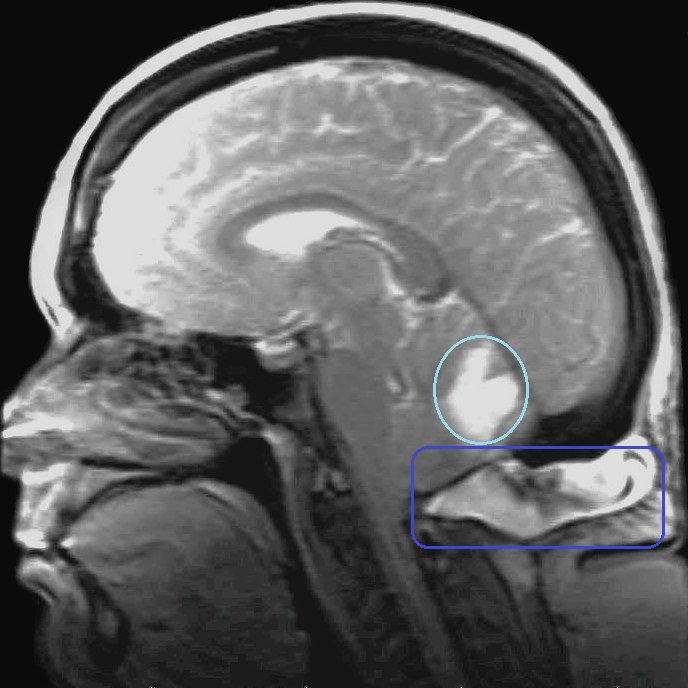
Post-op Complication: Pseudomeningocele
When I was released from the hospital following decompression surgery, I was instructed not to lift, push, or pull anything for two weeks so that my dura patch would have a chance to adhere. The problem was, I could feel fluid squeezing out of the patch far beyond that two-week limit. I developed a pseudomeningocele (blue box above), which can be normal immediately after surgery before the dura adheres, but as long as there is no active leak, the body should absorb the fluid and the pseudomeningocele should quickly resolve. My neurosurgeon tapped some of the fluid out with a syringe twice and we waited patiently to see if it would subside on its own. It did not subside and in December 2012 (just over two years post-decompression), I developed acute vertigo. Everything was spinning and rocking, non-stop. It didn’t matter if my eyes were open or closed. I was waking up vomiting in my sleep from the dizziness. I couldn’t walk at all without falling hard to my right. I had no sense of balance at all and it didn’t just come and go, it was constant. Another MRI was done and it showed that my cerebellum was absorbing the fluid from the pseudomeningocele (so the cerebrospinal fluid was inside my brain, not just surrounding it; see light blue circle in image above). The decision was made to put in a subgaleo-peritoneal shunt (SP shunt), which runs from the pseudomeningocele to my peritoneum. They expected that it might take up to six months to fully drain from my cerebellum, but I woke up from the anesthesia with no signs of vertigo. I believe this surgery saved my life, but as with all shunts (especially amongst those with EDS, which I had not yet been diagnosed with), the shunt was destined to cause problems all by itself.
My Many Shunt Revisions
In April 2013, an unrelated CT Scan revealed that my shunt was no longer in my peritoneum. My NS scheduled for a general surgeon to “tie in” my shunt so it would not happen again (surgery #3). We went several months without complication until that November. The tied in shunt pulled out of my peritoneum again (it was excruciating). Hoping gravity would help in the matter, my NS did an incision just under my right rib cage and dropped it down into my peritoneum (surgery #4). Shortly thereafter, radiologist reports started showing a concern for the location of my brain and I was diagnosed with “Sagging Brain Syndrome.” So my six-week post-op appointment (which my NS did faithfully after every surgery) became my pre-op appointment for my 5th related surgery. This time a non-adjustable valve was attached to the shunt (at my chest) in hopes that by slowing down the amount of CSF being drained by the shunt, my head could retain more fluid and my brain could once again lift and become buoyant. Five months later I developed a hernia and upon closer examination (during surgery), it was found that my peritoneum was literally falling apart from all the trauma of the shunts; so my hernia removal surgery became a reconstruction surgery where my abdominal wall was pulled together with mesh, while carefully ensuring that the shunt didn’t come out (surgery #6). The shunt never moved again. As my brain continued to sag, the choice was made to replace the valve with an adjustable valve and in November of that same year, I was having surgery #7. The valve was adjusted to its slowest possible setting in hopes of finding a balance where it drained enough to keep the hydrocephalus at bay, yet retain enough CSF to lift my brain and keep it lifted and out of my spinal canal (so we could establish flow to the spinal canal and avoid the possibility of a syrinx).
Diagnosis: Ehlers-Danlos Syndrome
Despite my concerns that I might have a connective tissue issue and being told over-and-over again that I “didn’t look like someone with Ehlers-Danlos Syndrome,” I was finally diagnosed with it in May 2015. After finally finding a neurologist who understood the role that our connective tissues can have in a Chiari Malformation, I was given a referral to a geneticist. It still wasn’t as easy as it should be though. The geneticist did not know much about Chiari or Ehlers-Danlos related conditions (although he didn’t initially admit to that), so I had no idea at that point what was and was not related, and neither did my doctors. I received a call from the geneticist’s assistant and I agreed to send her pictures of my hypermobile maneuvers from the Beighton Scale. I could do all but bend over and put my hands flat on the floor with my knees straight, but I was able to do that when I was younger (and thinner). I was given a 9/9 on the Beighton Scale and was told that he would just mark my chart as diagnosed “hypermobile” and that he didn’t need to see me. I honestly didn’t know any better at this point, but I was about to learn something very important. I sat there thinking about what this “hypermobile” diagnosis would mean for me and decided to look more into EDS for myself. I read about the high risk of aneurysms, organ tearing, miscarriages, etc. and I was back on the phone with that assistant within twenty minutes. She asked if she could call me back, and within the hour the geneticist had decided that he needed to see me. He set up an appointment with me within twenty-four hours and asked if it was okay if he had a few others (doctors and medical students) there as well, since they’re a training hospital and they “don’t really come across patients with Ehlers-Danlos” (he should have told me that from the beginning). I agreed. Despite his lack of knowledge on EDS related comorbidities, he did know exactly where on my body to look for characteristics of EDS (all of which I thought I didn’t have). For instance, my skin isn’t unusually elastic, except in my upper arms and upper thighs. My skin isn’t translucent (I’m olive complected), except for on my breasts, back, and inner forearms. My skin isn’t unusually soft, except on my back. Now concerned that I might have Vascular Type EDS (vEDS), he decided to have me tested for that. The test was easy on my part but expensive on theirs. They drew blood and had it refrigerated and shipped to a lab in Washington state. It took thirty days for them to make sure that there was no mutation in my COL3A1 (collagen 3; alpha 1) gene, which has a median mortality age of 48. Initially, I felt devastated, since I was already 44. I decided that I hadn’t fought through all that I had, to only live a few more years. Thirty days later, the test came back indicating that I didn’t have vEDS and by default, I was diagnosed with Hypermobility Type EDS (hEDS). I was relieved, but the geneticist assured me that I still needed to be cautious. Since EDS symptoms are known to cross the type boundaries, and we already knew that vascular complications ran in the family (with the aneurysms) and with me personally (my peritoneum tearing), it technically made me “hEDS with vEDS crossover symptoms” and I’d probably have to explain that to my doctors for the rest of my life, so they remain aware of my potential to have additional vascular problems.

My Poor Mess of a Neck
The electric shock feeling in my spine (Lhermitte’s Sign) that I’d had intermittently for years, became an all-day, everyday thing, and much stronger in intensity. The MRI revealed that the herniated disc I had between my C3/4 was getting worse. The disc was removed with cadaver put in its place and the discs were fused together. My 8th surgery (ACDF = Anterior Cervical Discectomy and Fusion) wasn’t related to Chiari, but it was related to the EDS. We knew that my cervical spine was really bad from the beginning, but it got worse. I am now actually diagnosed with Degenerative Disc Disease in all three levels of my spine, but my neck has by far taken the brunt of it all. The ACDF, while 100% necessary, compromised the discs adjacent to it, and every disc from C4-7 is either bulging or herniated (Subaxial (cervical) Instability), so additional surgeries are likely to be needed.
Learning to Advocate for Myself
Over the past several years I have become an enthusiast of Chiari related research and MRIs (out of medical necessity more than anything). It became apparent to me that I absolutely needed to know everything that was going on in my body in case my doctors didn’t. When I first started, I’d print out studies and lay in bed with multiple high-lighters. I had such brain fog that I’d lay there crying at the fact that I was reading and rereading the same paragraphs over again, but I knew that I had to learn it despite how impossible it seemed. I prayed a lot for God to help me with my understanding and He did. I also started looking at the medications I was taking, the supplements I was taking, and what the ideal doses were for me (especially those that would help with inflammation and cognition), and other natural remedies. The first thing that I removed was all of the nerve meds that they had me on for peripheral neuropathy. I was maxed out on Nortriptyline (a tricyclic antidepressant) and almost maxed on Gabapentin (both of which had caused me to gain an incredible amount of weight over the years). When I informed my primary care doctor that I wanted to go off of them all, he thought it was a bad idea because of the severity of my neuropathy. I insisted though and asked him to help me to wean myself off of both of them in healthy intervals, and let’s “just see.” With the first down-dose, I physically felt a reduction in inflammation. It took me many months to wean off and get them out of my system, but in hindsight, I think this was the single best decision that I could have made. The longer I was on supplements instead of the nerve meds, the more my brain-fog improved, and I now believe that I have regained all that I’ve lost cognitively and then some.

In 2016, I was reviewing some of my old MRIs and I saw a large CSF filled hole in my lower medulla oblongata (lower brainstem). It was obvious in all MRI series since 2015, yet I was told that all was stable. After researching it, I asked my neurologist to take a look and see if it could be Syringobulbia. She referred my question to my neurosurgeon and he confirmed that I had an 11mm cyst in my brain stem. This type of cyst happens when there is a blockage of cerebrospinal fluid and is most frequent when the brain stem is also herniated below the foramen magnum (Chiari 1.5). It explained a lot of the problems that I was having, that we had thought to be unrelated. For instance, and I had a decreased sensitivity to temperature for years, never feeling hot or cold; and never having the automatic reactions that I should have had in response to temperature, like sweating and shivering. I could comfortably be outside in heat above 100° without breaking a sweat, or be outside in shorts and a tank-top when it was a chilly 30° morning without ever shivering. I also developed tachycardia and I am now medicated to keep my heart rate down to a safe level. My neurosurgeon ordered a new MRI in April 2017. The size of the syrinx had decreased to 9mm but was draining down my spinal cord forming an additional syrinx (Syringomyelia).
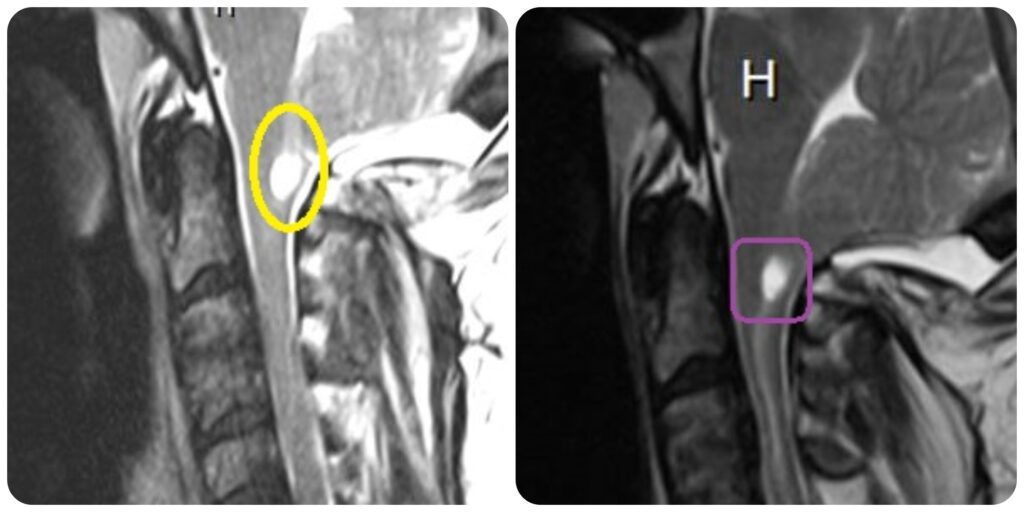
Consulting a Specialist
After all that I had been through in my fight, in April 2017, I decided to pay out of the pocket and have an online consultation with a Chiari Specialist in New York, who specializes in Chiari with EDS (the best $300 that I’ve spent in my fight). I sent him my pertinent medical records and copies of my MRIs in advance, wanting to find out what my doctor did right, and what he did wrong; and what course of action should be taken at that point. My expectation was that he would give me reasons why I should go to New York to see him, but that’s not at all what he told me. He told me what my doctor did right and that he didn’t disagree with the course of action that my neurosurgeon wanted to take. He said that my brain had sagged as low as it really could, but that since my high/low pressures had balanced out, and I was feeling better than I had in years, my syringes really should dictate our next course of action.
In March 2018, following an exceptional year (at least where my head and neck are concerned) new imaging was done. My neurosurgeon asked me to come in to review it. It gave me a chance to tell him about the specialist’s opinions. My MRI showed that the Syringobulbia had decreased another 2mm. I asked him what that meant for the cervical syrinx, and that had almost completely disappeared. I asked him to go back to my images and correct me if I was wrong, but “the only reason that a syrinx (in either location) would dissipate like that was if I was finally getting CSF flow down my canal (despite my severe brain sag).” He agreed and I think he was a little surprised to see me think on my feet and figure that out in front of him (where I wasn’t having to ask anyone or look it up). He also confirmed that I had an Acquired Chiari, secondary to Intracranial Hypertension. He applauded me for learning all that I had and said that he wished that he had checked my pressures before decompressing me, as it may have changed the course of action that we had taken. And we agreed to wait a year and see where the syringes (syrinxes) are. As I left his office that day, I felt such a sense of relief, that we were finally getting CSF flow like the decompression in 2010 was meant to do.
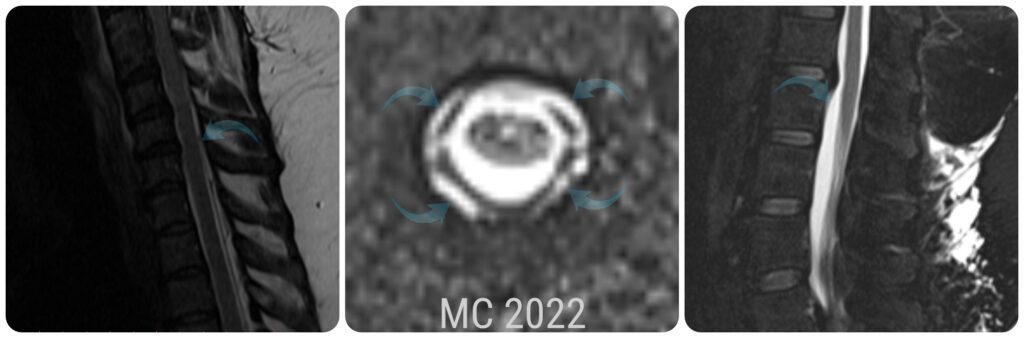
My Extensive Epidural CSF Collection
In 2022, my neurosurgeon contacted me telling me that he was retiring and he’d like to have one last MRI of my entire spine (he added the brain to the request at my request). Unbeknownst to me, he ordered a CSF Leak Protocol, which consists of less slices, but they’re specifically looking for leaks. The images showed an “extensive extradural CSF collection from C7-L4, consistent with a CSF Leak and probable dural tear or CSF Venous Fistula.” They followed up with a Dynamic CT Myelogram. A Dynamic is different than a regular CT Myelogram, as they do it over 2-3 days, and they insert the contrast little by little into my spinal canal, and watch carefully for it to leave the spinal canal. CSF leaks and dural tears aren’t uncommon amongst Ehlers-Danlos patients, and usually happen in the front or back of the canal. CSF Venous Fistulas on the other hand are a much newer phenomenon, and they usually happen on the sides of the canal (more often on the right side). After two days of grueling tests, they found no active leaks or evidence of fistulas and surmised that what they saw on the MRIs to be “residual artifacts” from a leak that I had in the past… a leak that could have pulled my brain down into my spinal canal in the first place.
It’s been a long road, hard road. I still battle inflammation and I’m definitely not done with surgeries. Eventually, I will need a ventriculoatrial (VA) shunt to hopefully resolve my high-pressure issues and enable us to remove my over-draining SP shunt that is making my brain sag. But for right now, I’m just enjoying feeling so much better! I praise God every step of the way, as I know that He’s there making a way out of no way. I have no idea why He took so long or why others haven’t seen the same results (because He loves them as much as He loves me), but I don’t have to have all the answers. I’ll just praise Him through the course of my journey, as He’s never let me go through it alone!
*I dedicate this story to my family: John (husband), Ron (dad), Johnathan (son), MyKaella (daughter), Jojo (son) and my daughters-in-law, Violet and Sarah. Thank you all for all your help and for standing and kneeling beside me throughout my entire ordeal. You’ve been there for me and loved me through this long haul and I praise God for each and every one of you.
Originally written in 2018. Updated April 2022.
Very educational while telling a personal fight; keep it up because things are changing for the Chiari community as well as the EDS community ❤
Its been a long fight but we got this mom.
Wow. A long, harrowing road. So many twists and turns but it’s obvious the right path is being taken! Thank you Michelle for your perseverance!
I so adore you! What an amazing journey of absolute fortitude and diligence! You have and will continue to inspire so many to fight….to learn and never give up. We have a great leader in you and a great example. Thank you love for sharing your journey and giving hope to so many!
Wow, this is an awesome read. Thanks for sharing your story. You’re truly an inspiration to me and I’m certain many others. Prayers to you regarding your healing and health.
Wow!! What a journey you’ve been on! Your story is such an example of why we must really research and advocate so fiercely for ourselves. Had you backed down or just accepted what doctors said, the results would’ve been very dire, I’m sure. I hope others reading this story can take inspiration from it to stay strong during their own battles!
You have had such a long, hard battle Sis but I 100% believe the research you do is what has ultimately saved your life. This story is going to help so many out there like you struggling along with surgery after surgery, without answers and worsening issues. We must fight to eduacate ourselves and our doctors to turn things around.